





Authors:
Jiangbin Zheng、Han Zhang、Qianqing Xu、An-Ping Zeng、Stan Z. Li
Paper:
https://arxiv.org/abs/2408.10247
Introduction
Enzymes, specialized proteins that act as biological catalysts, are pivotal in various industrial and biological processes due to their ability to expedite chemical reactions under mild conditions. Despite their significance, computational enzyme design remains in its infancy within the broader protein domain. This is primarily due to the scarcity of comprehensive enzyme data and the complexity of enzyme design tasks, which hinder systematic research and model generalization.
To address these challenges, the study introduces MetaEnzyme, a unified enzyme design framework that leverages a cross-modal structure-to-sequence transformation architecture. This framework aims to enhance the representation capabilities and generalization of enzyme design tasks, particularly under low-resource conditions. MetaEnzyme focuses on three fundamental enzyme redesign tasks: functional design (FuncDesign), mutation design (MutDesign), and sequence generation design (SeqDesign).
Related Work
The field of protein design has seen significant advancements with AI-driven approaches, particularly in inverse folding and conditional protein generation. Notable models include Structured Transformer, GVP-GNN, ProteinMPNN, PiFold, and ESM-IF, which have made strides in sequence recovery and structure prediction. However, enzyme engineering, especially in the context of few/zero-shot learning for mutation fitness, remains a niche domain with models like DeepSequence, ESM-1v, Tranception, and ESM-2 leading the way.
MetaEnzyme builds on these innovations by integrating learned structural and linguistic insights to address comprehensive design tasks, thereby enhancing functionality prediction and generalization across diverse proteins and functional enzymes.
Research Methodology
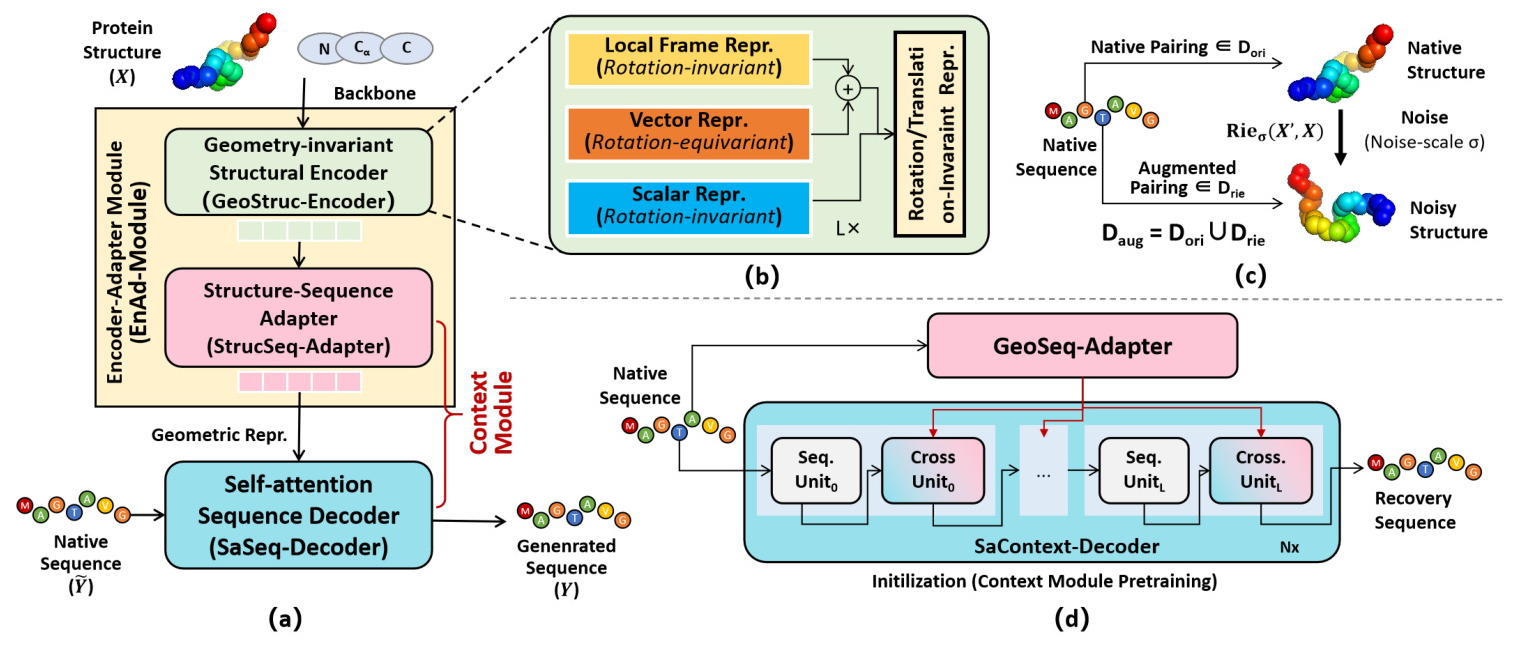
Universal Protein Design Network (UniProt-Net)
The core of MetaEnzyme is the Universal Protein Design Network (UniProt-Net), which consists of three main components:
- Geometry-invariant Structural Encoder (GeoStruc-Encoder): This module ensures rotation and translation invariance by using a lightweight GVP module that outputs rotation-equivariant vector features and rotation-invariant scalar features.
- Structure-Sequence Adapter (StrucSeq-Adapter): This adapter bridges the gap between structural and sequence representations, ensuring transformation equivariance.
- Self-attention Sequence Decoder (SaSeq-Decoder): This decoder incorporates fully self-attention layers to generate amino acid sequences conditioned on the protein backbones.
Energy-motivated Data Augmentation
To enhance structural representation, the study introduces an energy-driven Riemann-Gaussian geometric technique for data augmentation. This technique ensures that structural variations maintain the energy dynamics of proteins, preserving pairing relationships and geometric characteristics.
Context Module Initialization
The Context Module is initialized using a sequence-to-sequence recovery task in an autoencoder mode. This allows the module to acquire contextual semantic knowledge, utilizing cross-entropy loss for learning.
Experimental Design
Training Pipeline
The training pipeline involves initializing the parameters of UniProt-Net based on prior knowledge and incorporating Riemann-Gaussian data augmentation. The augmented dataset is used to train the model, with the protein backbone input into the EnAd-Module to produce geometric context features, which are then fed into the decoder to generate the sequence distribution logits.
Domain Adaptation for Low-Resource Enzyme Design Tasks
MetaEnzyme employs task-specific meta-learning and domain-adaptive techniques to address low-resource enzyme design tasks. The framework adapts to new tasks through a few gradient descent iterations, transforming the meta-learner into task-specific parameters.
Results and Analysis
Few-shot Learning for Function Prediction (FuncDesign)
MetaEnzyme demonstrates superior performance in fold-independent and fold-agnostic predictions, highlighting the significant improvement brought about by meta-finetuning. The model’s enhanced generalization ability is attributed to the incorporation of structural information.
Zero-shot Learning for Mutation Effects (MutDesign)
The novel mutation effect scoring method introduced in MetaEnzyme shows a significant advantage in terms of speed and accuracy. The model consistently achieves the best matching rank, implying that in-domain finetuning on enzyme data contributes to enhanced overall performance in mutation prediction.
Autoregressive Generalization for Sequence Generation (SeqDesign)
MetaEnzyme exhibits outstanding performance in sequence generation tasks, achieving impressive AAR scores across various datasets. The model’s ability to generalize to out-of-domain datasets further underscores its robustness and effectiveness.
Wet Lab Experimental Validation
To validate the reliability of MetaEnzyme, wet lab experiments were conducted on the reversible glycine cleavage system (rGCS). The results showed a notable Spearman’s rank correlation of 70.1% between predicted and experimental effects, reinforcing the practical efficacy of MetaEnzyme.
Overall Conclusion
MetaEnzyme represents a pioneering framework for enzyme design, offering a unified solution that enhances adaptability across various enzyme design tasks. Despite the resource-intensive nature of wet lab validation, the study demonstrates the potential of MetaEnzyme to transition from universal to unified enzyme design, enabling seamless adaptation through straightforward architectural modifications or lightweight adjustments.
Future work should focus on expanding datasets of functional enzymes, developing more robust models to enhance generalization, and conducting additional wet lab experiments to further validate the framework’s efficacy.

















