





Authors:
Dingyi Rong、Wenzhuo Zheng、Bozitao Zhong、Zhouhan Lin、Liang Hong、Ning Liu
Paper:
https://arxiv.org/abs/2408.06391
Introduction
Accurately predicting enzyme functions using Enzyme Commission (EC) numbers is a significant challenge in bioinformatics. This process is crucial for understanding catalytic mechanisms, substrate specificities, and potential applications in various industries. Traditional experimental methods for determining EC numbers are time-consuming and resource-intensive, making computational methods particularly important.
Traditional bioinformatics methods, such as sequence alignment-based approaches like BLASTp, rely heavily on pre-existing knowledge stored in databases. These methods struggle with novel proteins lacking close homologs and the complexity of protein evolution. Recent deep learning approaches offer promising alternatives but often rely solely on either protein sequence or structure information, each with its limitations.
To address these challenges, we introduce MAPred, a novel multi-modality and multi-scale model designed to autoregressively predict the EC number of proteins. MAPred integrates both the primary amino acid sequence and the 3D tokens of proteins, employing a dual-pathway approach to capture comprehensive protein characteristics and essential local functional sites. Additionally, MAPred utilizes an autoregressive prediction network to sequentially predict the digits of the EC number, leveraging the hierarchical organization of EC classifications.
Related Work
Sequence Similarity-based Methods
Sequence similarity-based methods compare a query protein sequence against a database of sequences with known functions to find matches with high sequence similarity. These methods rely on the quality of sequence alignment and database annotations. Recent developments have integrated traditional homology searches with more sophisticated algorithms, such as Hidden Markov Models (HMMs), to detect homologous protein families and predict their functions, including EC numbers.
Sequence-based ML Methods
Sequence-based machine learning methods use neural networks to capture patterns within amino acid sequences to predict EC numbers. Early attempts used Convolutional Neural Networks (CNNs) to capture local sequence motifs. Recurrent Neural Networks (RNNs), particularly Long Short-Term Memory (LSTM) networks, were later explored for their ability to capture long-range dependencies in sequences. Recently, Transformer-based methods have achieved state-of-the-art performance by leveraging self-attention mechanisms to capture complex relationships between amino acids.
Structure-based ML Methods
Structure-based methods use the protein’s structure as input to capture interactions between amino acids. CNNs have shown promising results in capturing intricate patterns within protein structures, typically representing the input structure as Grids or Residue Contact Maps. Graph Neural Networks (GNNs) have also been explored, representing protein structures as graphs to capture relational information within proteins.
Methodology
Overall Framework
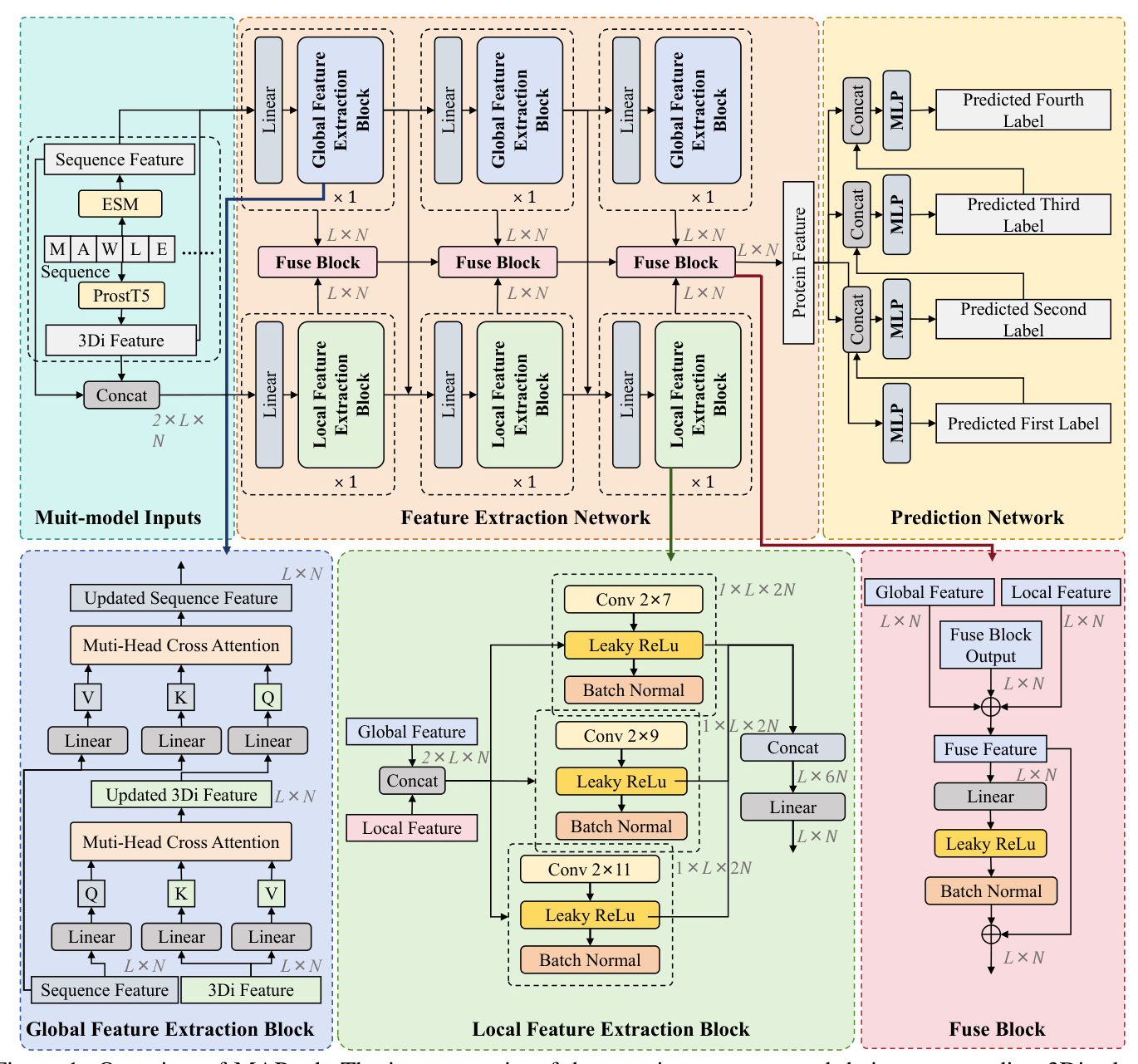
MAPred’s overall model framework consists of two networks: the Feature Extraction Network and the Prediction Network. The Feature Extraction Network includes a global feature extraction pathway and a local feature extraction pathway. The Prediction Network operates on an autoregressive prediction scheme, predicting the digits of the EC number sequentially.


Feature Extraction Network
Global Feature Extraction Block
The Global Feature Extraction (GFE) pathway uses a strided sequence-to-3Di cross-attention mechanism to integrate protein sequence features with their corresponding structural features. Each GFE block consists of two cross-attention layers, allowing for a comprehensive integration of both sequence and structural information.

Local Feature Extraction Block
The Local Feature Extraction (LFE) pathway employs a CNN-based approach to construct contextual features for each amino acid, enhancing the precision of functional site identification. The LFE pathway consists of three LFE blocks, each containing convolution kernels of different sizes to extract both short-range and long-range dependencies within the protein sequence.

Prediction Network
The Prediction Network uses an autoregressive prediction strategy to progressively predict each digit of the EC number. A multi-task learning framework comprising four MLPs is designed, each responsible for predicting one digit. The input features for each MLP include both the fused features and the prediction results from the previous MLP.
Model Training
Training Loss
A combined training loss is utilized, consisting of the triplet loss between samples of different classes and the BCE loss between the ground-truth and predicted EC number.
Two-phase Training
A two-phase training scheme is adopted. In the first phase, the feature extraction network is trained using triplet learning. In the second phase, the EC number prediction network is trained, with the feature extraction network frozen.

Experiments
Experimental Protocol
Datasets
The Swiss-Prot dataset and data splitting used in CLEAN are employed, encompassing 227,362 protein sequences covering 5,242 EC numbers. The performance of MAPred is evaluated on real-world datasets, including New-392, Price-149, and New-815.
Implementation Details
In the first training phase, the feature extraction network is trained with a batch size of 40 for 1000 epochs. In the second phase, the EC number prediction network is trained with a batch size of 50000 for 150 epochs. The Adam optimizer and CosineAnnealingLR scheduler are used. Experiments are conducted on a machine with three NVIDIA RTX6000 ADA GPUs.
EC Number Prediction
MAPred demonstrates remarkable performance, outperforming existing approaches in all evaluated metrics. On the New-392 dataset, MAPred achieves a Precision of 0.651, a Recall of 0.632, and an F1 score of 0.610. Consistent performance gains are observed across other datasets as well.

Ablation Studies
Component contribution analysis experiments verify the effectiveness of MAPred’s components. The global feature extraction pathway, local feature extraction pathway, and autoregressive label prediction model each play significant roles in the model’s performance. Multi-modality inputs also enhance results.

MAPred Learns the Functional Regions of Enzymes
MAPred can classify enzymes based on their EC numbers by extracting latent features from amino acid sequences. Attention scores computed in the attention layer are visualized on the three-dimensional structure of the enzyme, revealing that MAPred primarily concentrates on catalytic sites where reactions occur.
[illustration: 7]
Conclusion
MAPred is a novel approach to enzyme function prediction that integrates both sequence and structural data. The model’s performance surpasses existing methods, achieving high F1 scores on various test sets. Extensive ablation studies validate the effectiveness of each component within MAPred. Future work will focus on excavating key protein regions and incorporating more diverse datasets, expanding the prediction of labels to Gene Ontology (GO) terms.


