

Authors:
Tianyu Zhang、Yuxiang Ren、Chengbin Hou、Hairong Lv、Xuegong Zhang
Paper:
https://arxiv.org/abs/2408.10124
Introduction
Molecular property prediction is a cornerstone of drug discovery, enabling the identification and optimization of potential drug candidates. Traditionally, this task has relied heavily on biochemical expertise and extensive domain knowledge, which is both time-consuming and costly to acquire. Recent advancements in deep learning, particularly the use of pre-trained models, have shown promise in automating and enhancing molecular property prediction. However, these models often require large amounts of labeled data and domain-specific knowledge, posing significant challenges.
Large Language Models (LLMs) have demonstrated remarkable capabilities in understanding and generating general knowledge efficiently. However, they sometimes produce hallucinations and lack precision in domain-specific contexts. On the other hand, Domain-specific Small Models (DSMs) are adept at calculating precise molecular metrics but lack the breadth of knowledge required for comprehensive representation learning. To address these limitations, the study proposes a novel framework called MolGraph-LarDo, which integrates LLMs and DSMs for molecular graph representation learning.
Related Work
Molecular Representation Learning
Molecular representation learning has evolved significantly, with various pre-training frameworks enhancing the accuracy and efficiency of molecular property prediction. Notable methods include:
- GROVER: Utilizes message-passing networks with a Transformer-style architecture for pre-training on unlabeled molecular data.
- GraphMVP: Employs self-supervised and pre-training strategies using 3D geometric information.
- PhysChem: A deep learning framework that incorporates external physical and chemical information.
- MG-BERT: A BERT-based pre-training framework leveraging large amounts of unlabeled data.
- MolCLR: A self-supervised framework that uses contrastive learning with Graph Neural Networks (GNNs).
These methods, while effective, often require extensive domain-specific knowledge and large datasets, which can be expensive and time-consuming to obtain.
Large Language Models for Molecular Tasks
Recent studies have explored the integration of LLMs into molecular tasks, leveraging their ability to generate and classify molecular properties. Notable examples include:
- GPT-MolBERTa: A BERT-based model that predicts molecular properties using textual descriptions generated by ChatGPT.
- LLM4mol: Enhances molecular property prediction by combining in-context classification results with new representations generated by LLMs.
- MolTailor: Optimizes representations by emphasizing task-relevant features with virtual task descriptions generated by LLMs.
Despite their potential, these methods often suffer from hallucinations and lack precision in generating domain-specific knowledge.
Research Methodology
Overview of MolGraph-LarDo
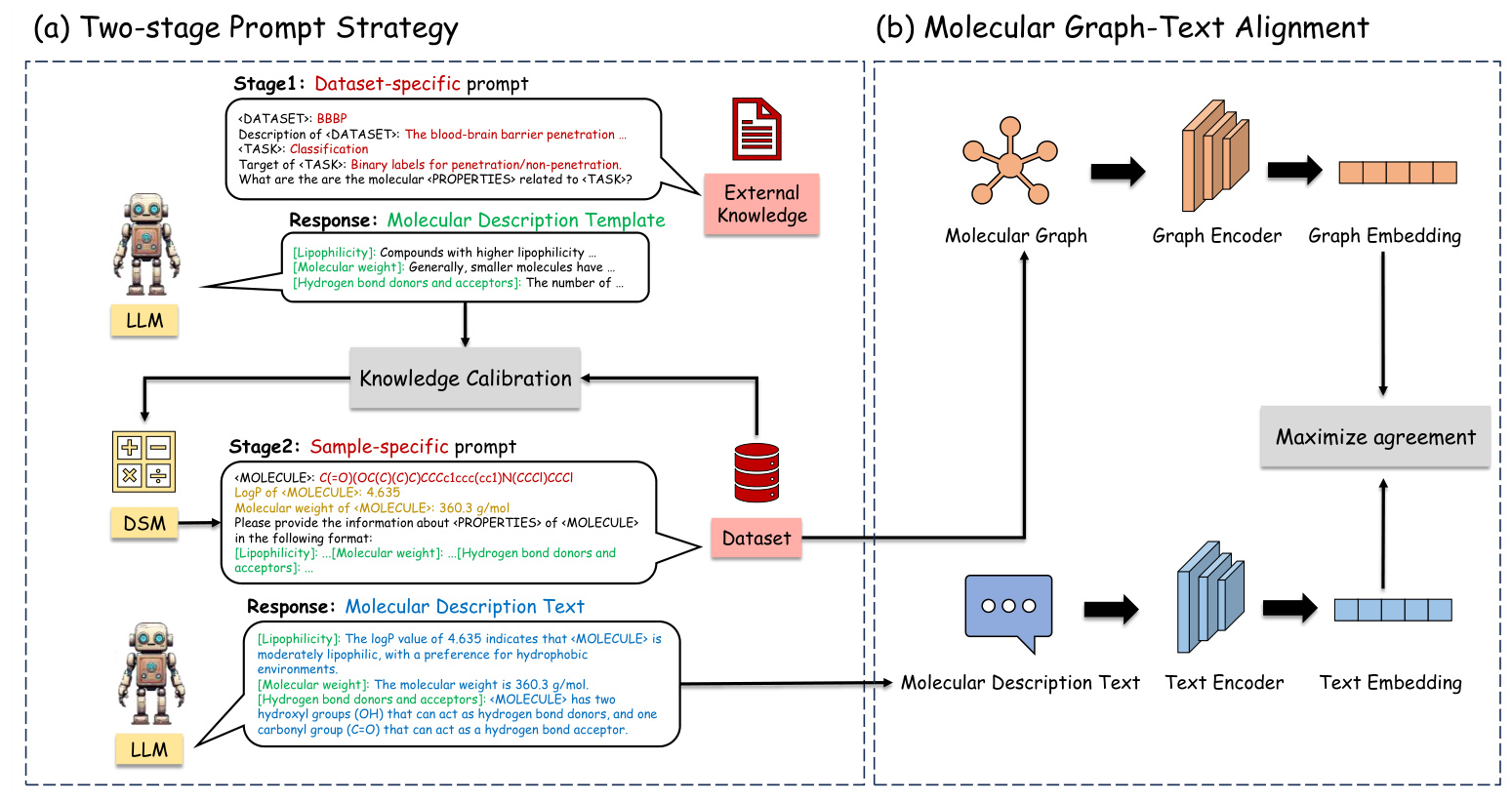
The proposed MolGraph-LarDo framework integrates LLMs and DSMs through a two-stage prompt strategy and a multi-modal graph-text alignment method. The framework aims to leverage the strengths of both LLMs and DSMs to enhance molecular representation learning.
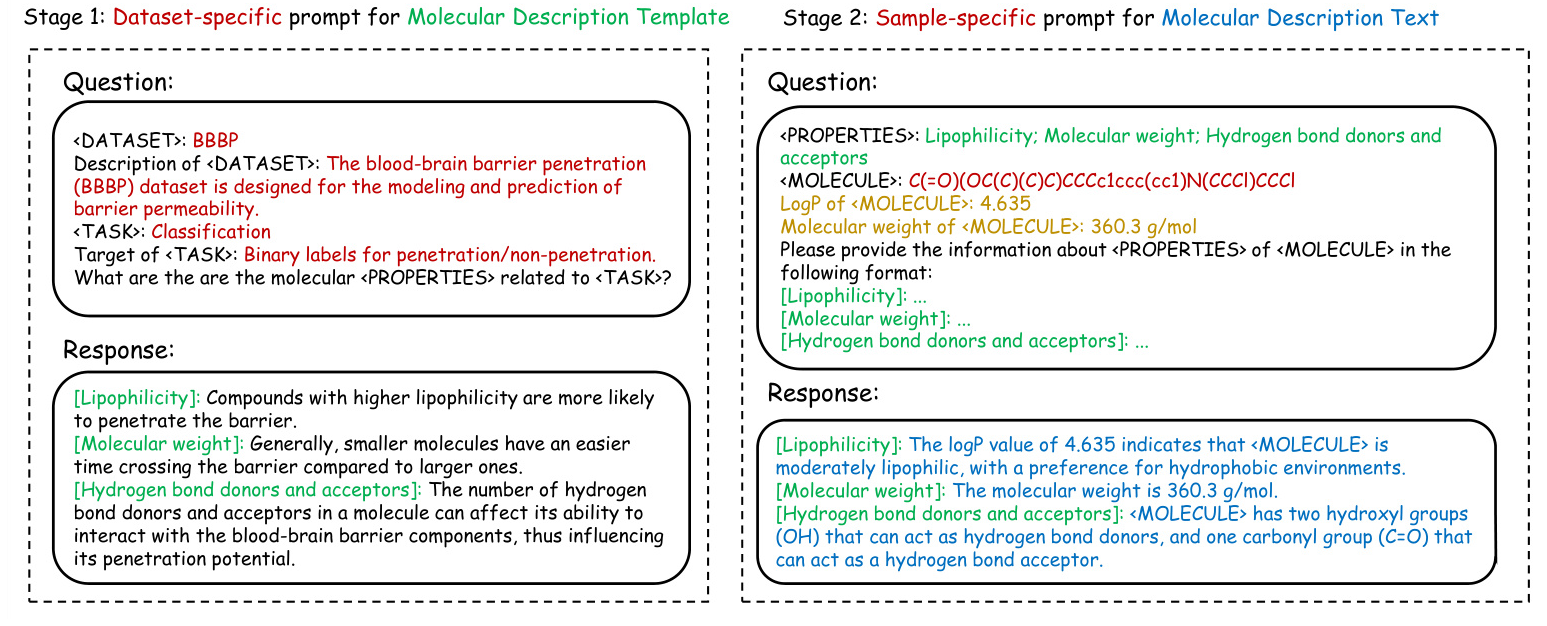
Two-stage Prompt Strategy
- Stage 1: Dataset-specific Prompt for MD-Template Generation
- A dataset-specific prompt is designed to generate relevant molecular properties, termed the Molecular Description Template (MD-Template).
-
The DSM is used to implement knowledge calibration, ensuring the accuracy of domain-specific metrics.
-
Stage 2: Sample-specific Prompt for MD-Text Generation
- The MD-Template and calibrated knowledge are incorporated into a sample-specific prompt to generate Molecular Description Text (MD-Text) for each molecule.
- The MD-Text provides detailed molecular information, enhancing the subsequent graph contrastive learning process.
Molecular Graph-Text Alignment
A multi-modal alignment method is employed to align the molecular graph with its corresponding description text. This alignment enhances contrastive learning by optimizing the agreement between graph and text embeddings, ultimately improving the performance of downstream molecular tasks.
Experimental Design
Datasets
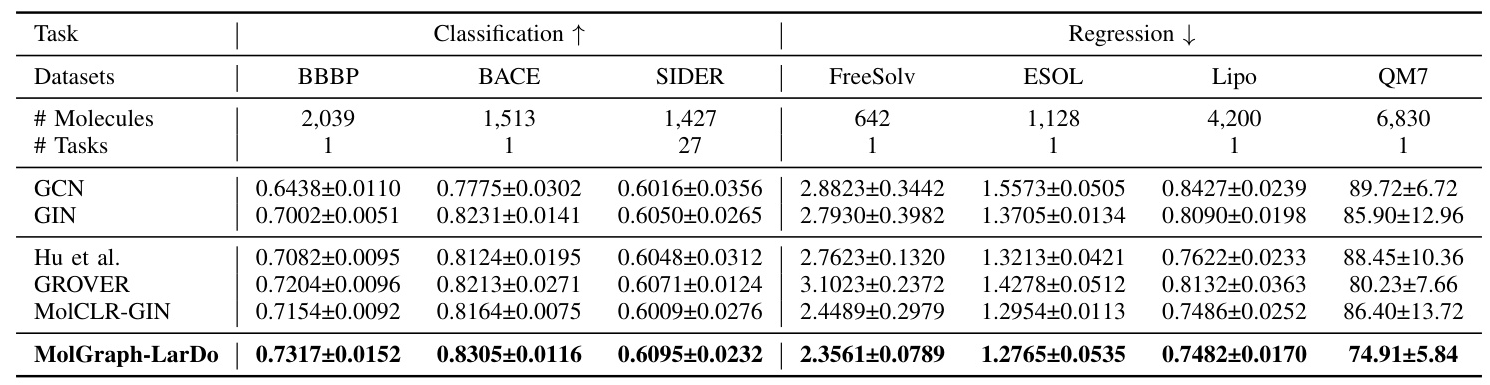
The proposed MolGraph-LarDo framework is evaluated on seven datasets from MoleculeNet, categorized into classification and regression tasks:
- Classification Datasets:
- BBBP: Tracks whether a molecule can penetrate the blood-brain barrier.
- BACE: Contains binary labels for molecules that inhibit human β-secretase 1.
-
SIDER: Includes data on approved drug molecules with side effects categorized into system organ classes.
-
Regression Datasets:
- FreeSolv: Includes hydration free energies of small molecules in water.
- ESOL: Records the water solubility of molecules.
- Lipo: Measures the lipophilicity of drug molecules.
- QM7: Derived from the GDB-13 database, used for studying molecular structures and their atomization energies.
Baselines
The performance of MolGraph-LarDo is compared with five baseline methods, including supervised methods (GCN, GIN) and pre-training methods (Hu et al., GROVER, MolCLR).
Implementation Details
- Graph Encoder: GIN with five layers and 256 hidden dimensions.
- Text Encoder: Sentence-BERT with frozen weights except for the last output linear layer.
- LLM: Mistral-7B-Instruct-v0.2.
- DSM: RDKit-2023.3.2.
The datasets are split into training, validation, and test sets using a scaffold split method. The pre-training stage uses Adam optimizer with a learning rate of 0.005, and the fine-tuning stage searches the learning rate within [0.0001, 0.0005].
Results and Analysis
Molecular Property Prediction
MolGraph-LarDo consistently outperforms all baseline methods across the seven datasets, demonstrating its effectiveness in enhancing molecular property prediction without the need for extensive domain-specific data.
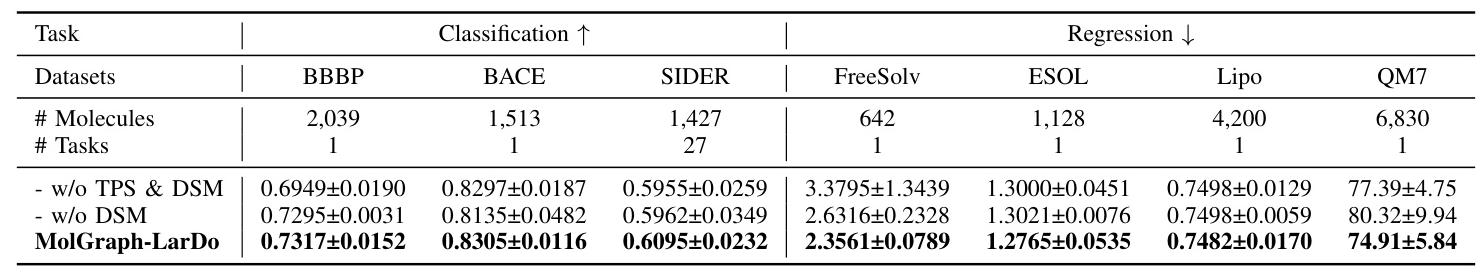
Ablation Study: Two-stage Prompt Strategy and DSMs
The ablation study shows that both the two-stage prompt strategy and DSMs significantly contribute to the performance of MolGraph-LarDo, justifying their inclusion in the framework.
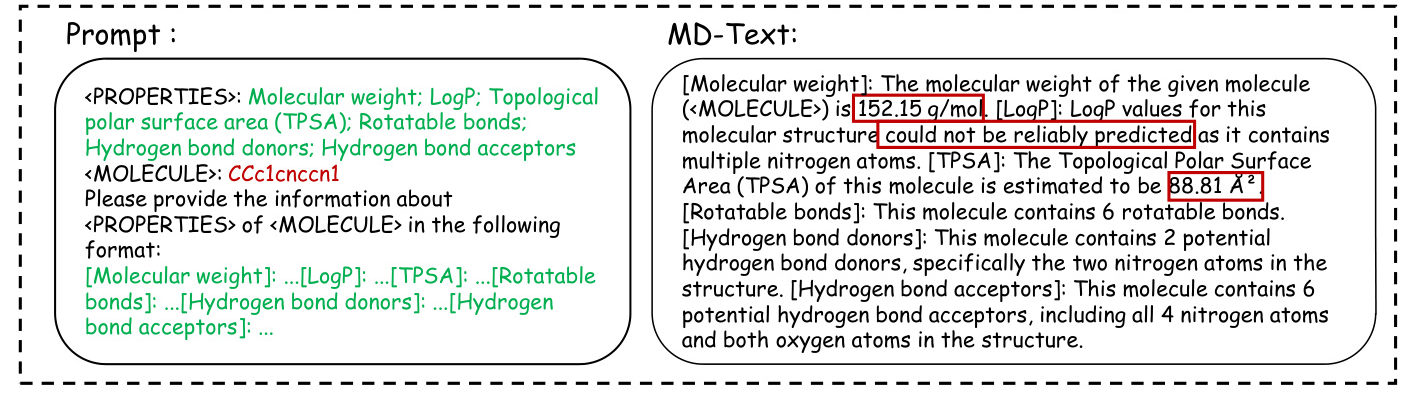
Case Study of MD-Text
The case study highlights the differences between the three versions of MolGraph-LarDo, demonstrating the importance of the two-stage prompt strategy and DSMs in generating accurate and relevant molecular descriptions.

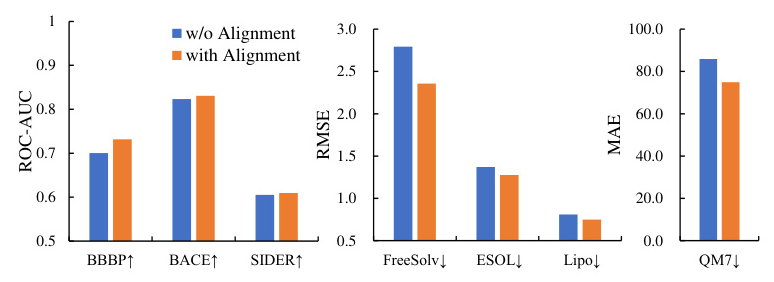
Graph-Text Alignment
The graph-text alignment mechanism effectively enhances the performance of MolGraph-LarDo, as evidenced by the ablation study results.
Overall Conclusion
The proposed MolGraph-LarDo framework effectively integrates LLMs and DSMs to enhance molecular graph representation learning. By leveraging the strengths of both models, MolGraph-LarDo addresses the limitations of existing methods, providing accurate and efficient molecular property predictions. The framework’s success is attributed to the innovative two-stage prompt strategy and the multi-modal graph-text alignment method, which together ensure the relevance and accuracy of the generated molecular descriptions. The results demonstrate that MolGraph-LarDo not only improves performance but also reduces the cost of acquiring domain-specific knowledge, making it a valuable tool for drug discovery and other molecular tasks.

Code:
https://github.com/zhangtia16/molgraph-lardo