

Authors:
Donghai Fang、Fangfang Zhu、Dongting Xie、Wenwen Min
Paper:
https://arxiv.org/abs/2408.06377
Introduction
In the realm of complex organisms, cells form specialized clusters through dynamic interactions and intricate organizational structures. These clusters coordinate the functions of the organism through mutual influences and tight connections. The latest Spatial Resolved Transcriptomics (SRT) technologies, such as ST, 10x Visium, and Stereo-seq, can comprehensively measure transcriptional expression at specific spatial locations (spots) while preserving the spatial context of the tissue. The key computational tasks in SRT data analysis are identifying shared and specific clusters, known as spatial domains, and addressing data denoising issues.
Traditional non-spatial clustering methods have failed to effectively utilize spatial information, resulting in incoherent clustering results within tissue sections. Recently, proposed spatial clustering methods consider the similarity between neighboring points to reveal the spatial dependence of gene expression. However, these methods overlook the supervisory signals in the latent space and the complex graph self-supervised contrastive learning frameworks lack consideration of global semantic information and the similarity of spatial structures.
To address these challenges, we propose a Contrastively Augmented Masked Graph Autoencoder (STMGAC) to learn low-dimensional latent representations for SRT data analysis. This method uses a masked graph autoencoder to reconstruct raw gene expression and perform gene denoising. Persistent and reliable latent space supervision information is obtained through self-distillation, guiding the latent representation for self-supervised matching and resulting in more stable low-dimensional embeddings. Additionally, positive and negative anchor pairs are constructed using triplet learning to augment the discriminative ability.
Proposed Methods
Overview of the Proposed STMGAC
In data processing, the raw gene expression data is divided into a mask matrix and a visible matrix. A mask GAE is used to reconstruct the raw data, and latent space supervision signals are obtained through a momentum encoder for matching latent representations. Selected anchor spots are then employed for triplet learning. The learned latent representations from STMGAC will be used in downstream analyses such as clustering and visualization 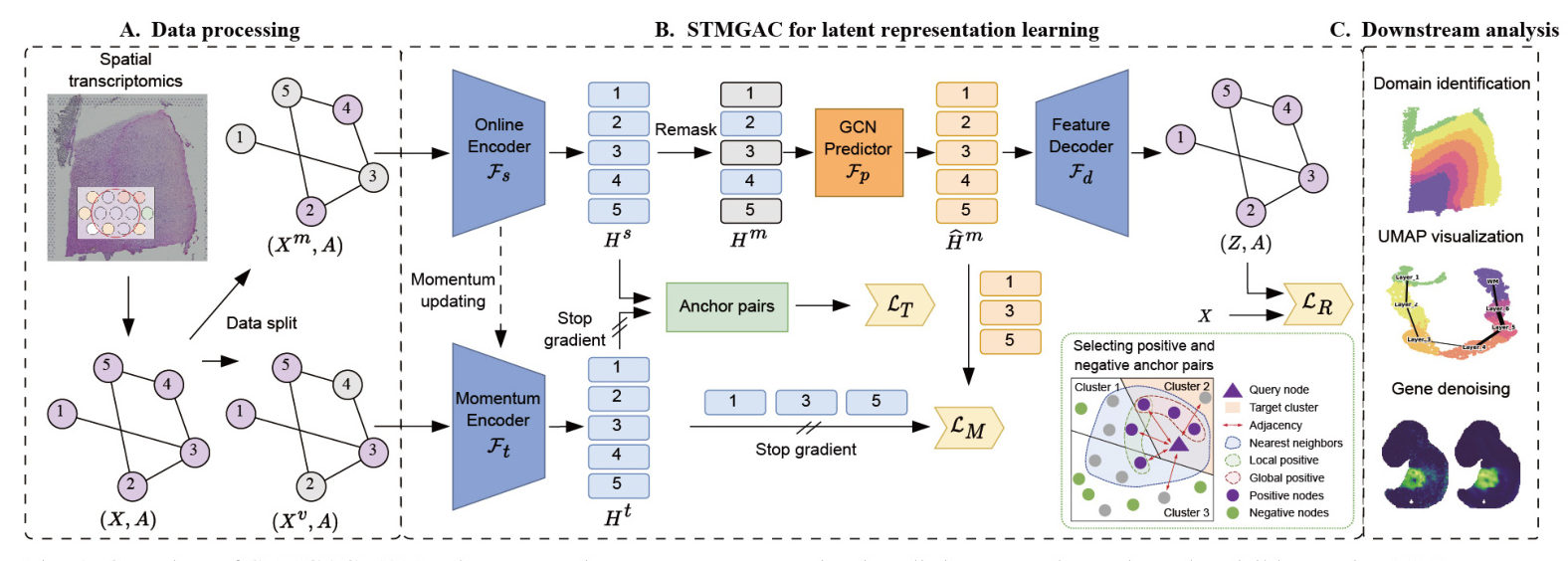 .
.
Data Preprocessing and Spatial Graph Construction
STMGAC uses gene transcription expression from SRT data and the spatial coordinates of spots as input. Using the library functions provided by SCANPY, the genes of interest are retained and the entire expression is log-normalized. The top Ng highly variable genes are retained to obtain the preprocessed data. The Euclidean distance between spots is calculated based on their spatial coordinates, and the K-nearest neighbors (KNN) algorithm is applied to select the K nearest spots, thereby constructing the adjacency matrix.
Data Augmentation with Spot Masking
Before training STMGAC, two complementary masked gene expression graphs are generated. They are used as the input for the online encoder and momentum encoder, respectively. With a masking rate ρ, a masked subset Vm is randomly sampled from the set of all spots in the SRT data, while the remaining spot set forms the visible subset Vv. The masked matrix and the complementary visible matrix are constructed to ensure that the spots masked in the latent space have persistent supervision signals and perform latent feature matching.
Latent Representation Learning with Masked Reconstruction
Graph Encoding
The online encoder Fs converts the masked matrix into a low-dimensional latent representation. An MLP composed of stacked linear layers is used for initial dimensionality reduction, followed by a graph encoder consisting of two layers of GCN to learn an effective latent representation from the visible spot neighbors.
Latent Representation Predicting
The representation predictor Fp obtains the predicted expression of the latent representation. The remask technique is used to mask the representation of the spots in the masked subset in the latent space, obtaining the remasked representation. A graph neural network allows the masked spots to learn the current features from the visible spots, and the final predicted latent feature representation is used to reconstruct the transcription expression of the raw data space and align with the supervision signal obtained by the momentum encoder.
Feature Decoding
An asymmetric autoencoder with a single-layer GCN is used as the decoder to map the predicted latent representation into the raw data space, obtaining the reconstructed gene expression matrix.
Reconstruction Loss in the Raw Data Space
The objective of STMGAC is to reconstruct the masked gene expression of spots in Vm given a partially observed spot set and adjacency relationships. The Scaled Cosine Loss (SCE) is used as the objective function to reduce the contribution from simple samples during the training process.
Latent Representation Matching for Robustness Enhancement
Momentum Graph Encoding
A momentum graph encoder with the same architecture as the graph encoder is responsible for encoding the raw gene expression data in the masked visible set to obtain a persistent momentum latent representation. This representation provides stable guidance for the latent representation. The parameters of the momentum encoder are updated by exponential moving average (EMA).
Matching Loss in the Latent Representation Space
The matching loss minimizes the distance between the predicted latent representation and the latent representation calculated by the momentum graph encoder, focusing on feature-level similarity.
Spot Triplet Learning with Positive and Negative Anchors
Selecting Positive and Negative Anchor Pairs
For a given query spot, the cosine similarity between the spot and all other spots is calculated. The k-nearest neighbor set in the latent space is used as a positive anchor candidate. The local positive anchor set and the global positive anchor set are defined based on neighboring information and global semantic spot set, respectively. The real positive set considers both local and global information. The negative candidate set is obtained by randomly selecting elements from the remaining spots.
Triplet Loss Enhances Discriminative Ability
Triplet loss encourages similar instances to be closer in the latent space while pushing dissimilar instances further apart, significantly improving spatial domain recognition capabilities.
Evaluation Criteria
Accuracy metrics such as Adjusted Rand Index (ARI), Normalized Mutual Information (NMI), Homogeneity (HOM) score, and Completeness (COM) score are used to describe the clustering precision of the method. The overall accuracy score is calculated as the average of NMI, HOM, and COM scores.
Experiments
Dataset Description
The clustering performance of STMGAC was analyzed on a range of SRT datasets from different platforms, including the human dorsolateral prefrontal cortex (DLPFC), human breast cancer (BRCA), anterior mouse brain tissue (MBA), human melanoma (HM), and mouse embryo (ME) datasets 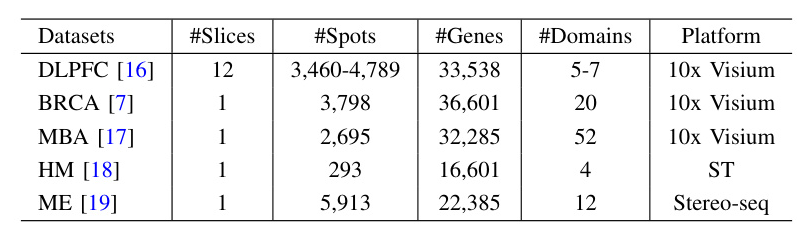 .
.
Baseline Methods
Several state-of-the-art methods were selected as baselines, including SpaGCN, CCST, DeepST, SEDR, STAGATE, GraphST, and DiffusionST.
Implementation Details
For STMGAC, a learning rate of 0.001 and a weight decay of 2e-4 were used, optimized with Adam. The online and momentum encoders had linear layers of dimensions 64 and 32, followed by GCN layers with output dimensions of 64 and 16. The predictor dimension was 32, and the feature decoder reconstructed to the raw data space. The default masking rate was 0.5. For anchor pairs selection, the top 50 nearest neighbors were used for the 10x Visium and Stereo-seq datasets, while the top 30 nearest neighbors were used for the ST dataset.
STMGAC Enables the Identification of Tissue Structures from SRT Data
Applying STMGAC to the DLPFC Dataset
STMGAC achieved the highest median ARI and ACC values across 12 slices of the DLPFC dataset, outperforming existing benchmark methods. The spatial domain identification results for slice 151675 showed that STMGAC correctly identified all layers with clear boundaries and no mixed spots. The UMAP visualization results indicated that the embeddings of STMGAC were consistent with the development trajectory of the layers 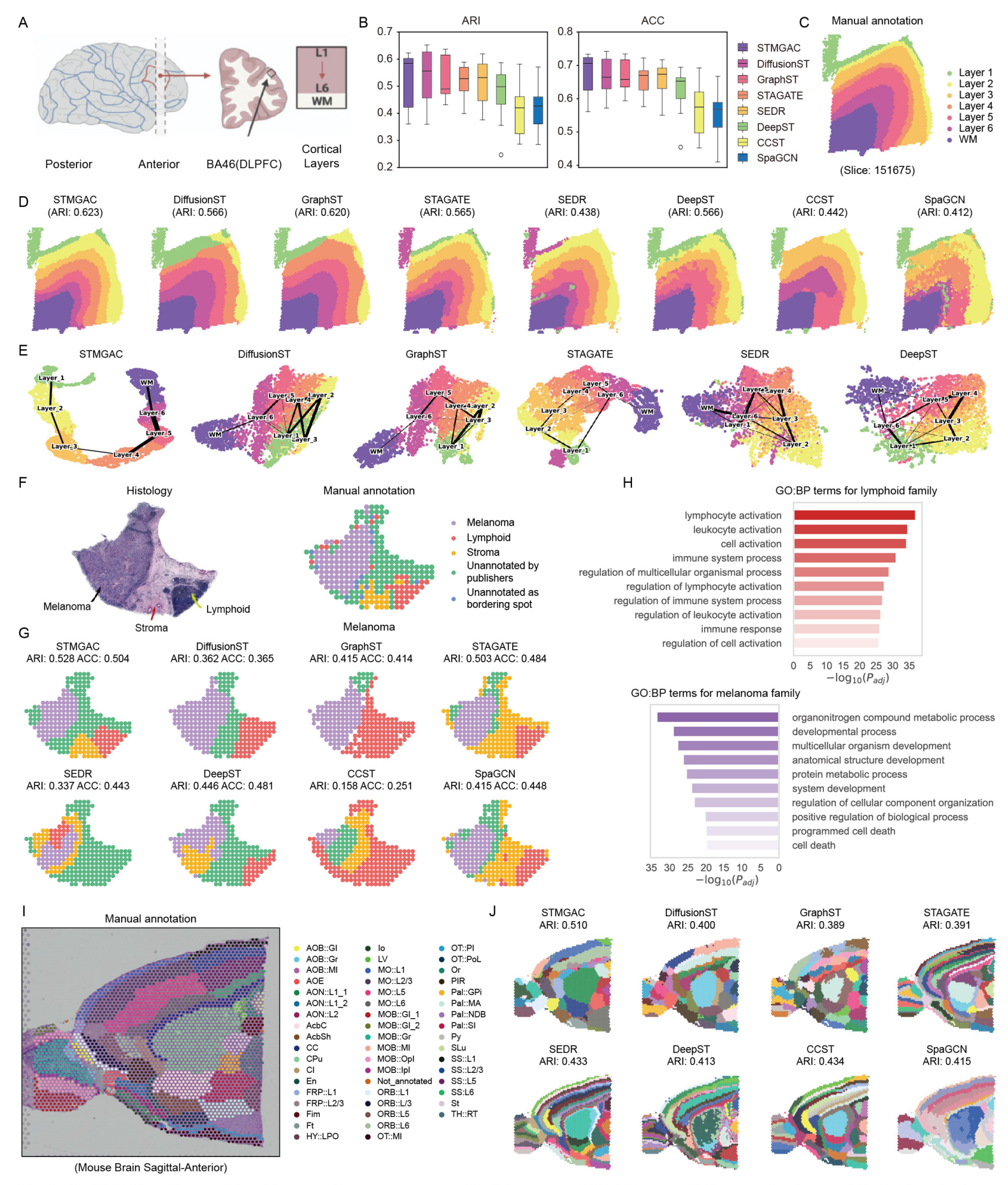 .
.
Applying STMGAC to the HM Dataset
In the HM dataset, STMGAC successfully identified the stroma region, which was missed by baseline methods. Differential expression analysis and gene enrichment analysis revealed key roles of identified genes in various physiological and pathological processes .
Applying STMGAC to the MBA Dataset
STMGAC demonstrated an improved ability to identify spatial domains in the complex tissue structure of the MBA dataset, outperforming other methods .
STMGAC Denoises Gene Expressions for Better Characterizing Spatial Expression Patterns
Applying STMGAC to the BRCA Dataset
STMGAC reduced noise in the BRCA dataset, better displaying the spatial patterns of genes. Differentially expressed genes in the tumor edge and health domains were identified, and their spatial expression was significantly enhanced post-denoising 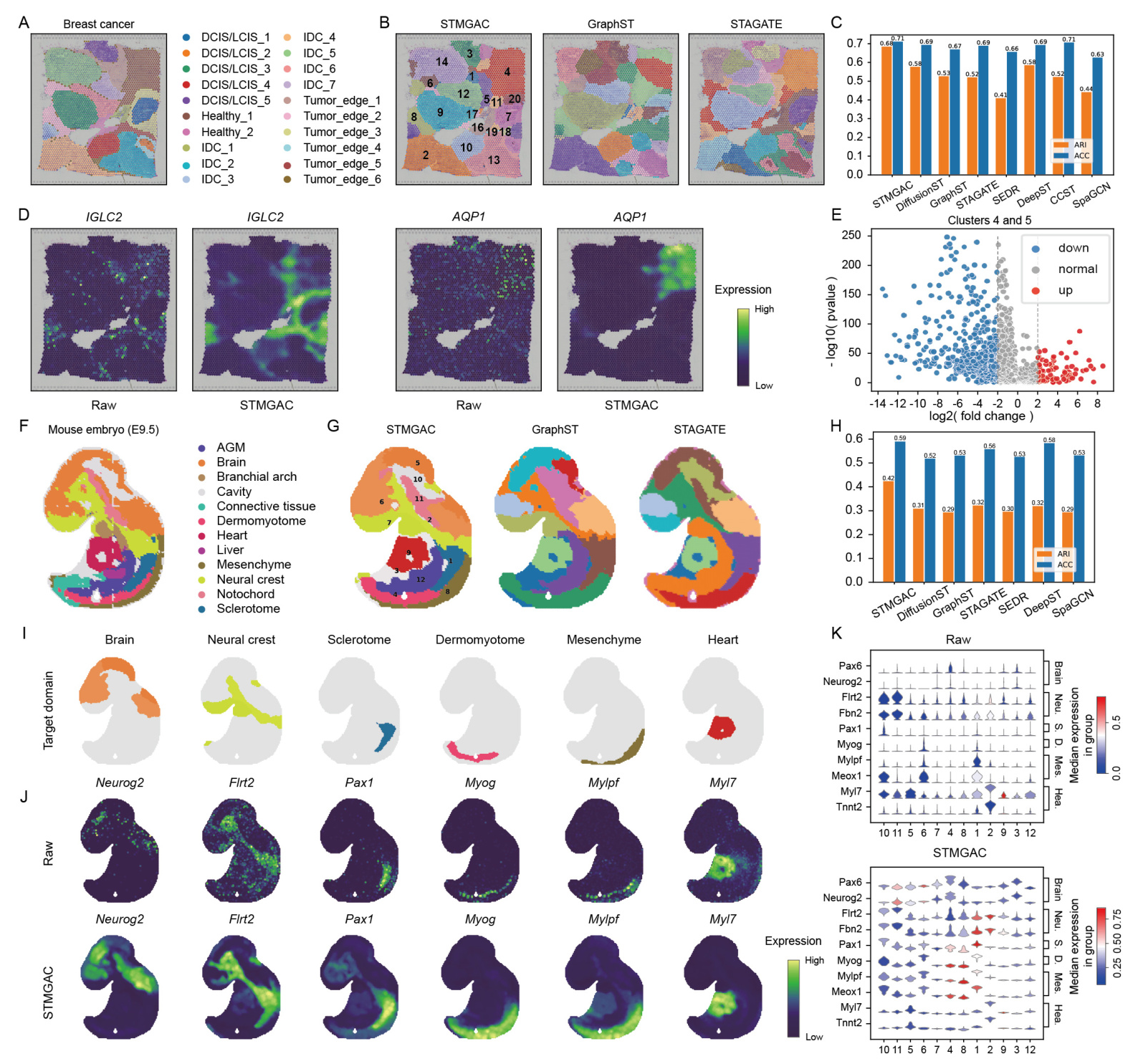 .
.
Applying STMGAC to the ME Dataset
The denoising capability of STMGAC was evaluated on the ME dataset by comparing the expression of key marker genes across tissue regions. The results demonstrated significant spatial enrichment of gene expression post-denoising, consistent with known functions .
Ablation Studies
Contributions of Different Components
Several variants of STMGAC were designed to investigate the contributions of different components. The results demonstrated that STMGAC achieved the best performance across multiple datasets, with slight underperformance on the sparse and low-density HM dataset due to the small number of spots 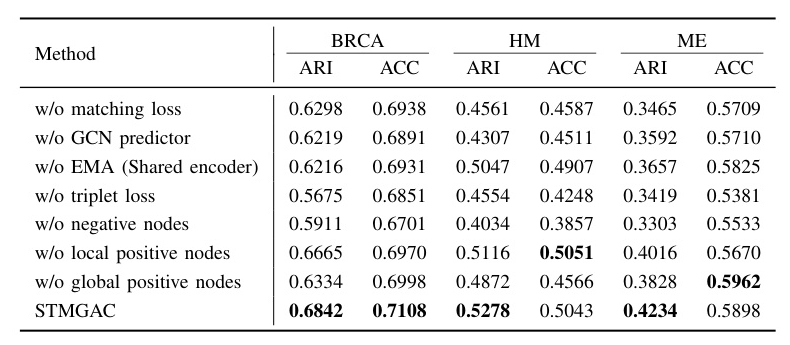 .
.
Impact of Different Loss Functions
The impact of reconstruction loss in the raw space, representation matching loss in the latent space, and contrastive loss was investigated. The results indicated that using SCE in the raw space and MSE in the latent space for mask matching made significant contributions, while TRI greatly enhanced clustering effects 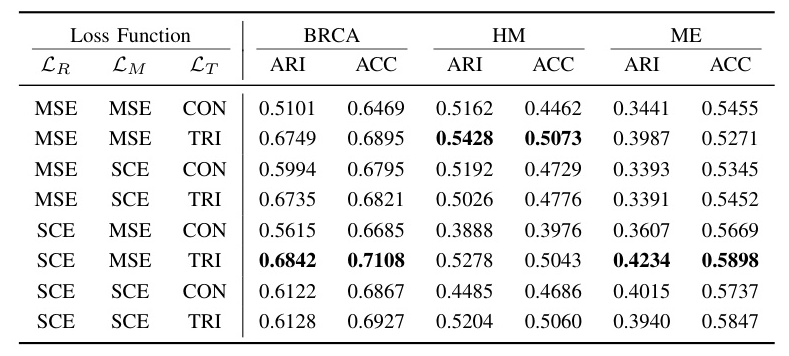 .
.
Conclusions and Discussion
In this paper, we propose a Masked Graph Autoencoder with Contrastive Augmentation (STMGAC) method for clustering and gene denoising analysis of SRT data. Previous graph masking methods did not focus on the supervision signals in the latent space, making it difficult for autoencoders to reconstruct raw data accurately. To address this, we designed the raw gene expression data as a masked matrix and a visible spot matrix, utilizing an EMA mechanism to provide persistent and reliable supervision signals for the model’s representation in the latent space. Furthermore, in the latent space, we select positive and negative anchor pairs based on the correlation between representations, local adjacency relationships, and global information, bringing similar instances closer together and pushing dissimilar instances further apart, thereby effectively enhancing spatial domain recognition capabilities. We analyzed the performance of STMGAC on datasets from different platforms and achieved results superior to the existing seven baseline methods. Additionally, ablation studies fully explored the contributions of each component to STMGAC.
Acknowledgment
The work was supported in part by the National Natural Science Foundation of China (62262069), in part by the Yunnan Fundamental Research Projects under Grant (202201AT070469), and the Yunnan Talent Development Program – Youth Talent Project.