

Authors:
Imran Nasim、Joaõ Lucas de Sousa Almeida
Paper:
https://arxiv.org/abs/2408.10720
Introduction
Scientific Machine Learning (SciML) has emerged as a transformative approach in traditional engineering industries, enhancing the efficiency of existing technologies and accelerating innovation. One of the critical areas where SciML has shown significant promise is in modeling chemical reactions. Chemical reactions are fundamental across various industries, providing insights crucial for innovation, product quality, and environmental management. However, modeling these reactions is non-trivial, often requiring the solution of stiff ordinary differential equations (ODEs) within Computational Fluid Dynamics (CFD) simulations. Stiff chemical kinetics are computationally expensive to solve within CFD simulations, prompting the exploration of various deep learning-based techniques.
In this study, we propose a novel approach utilizing a multi-layer-perceptron mixer architecture (MLP-Mixer) to model the time-series of stiff chemical kinetics. We evaluate this method using the ROBER system, a benchmark model in chemical kinetics, to compare its performance with traditional numerical techniques. This study provides insight into the industrial utility of the recently developed MLP-Mixer architecture to model chemical kinetics and motivates its use as a base for time-series foundation models.
Related Work
The challenge of solving stiff chemically reacting problems within the CFD framework has led to the development of various deep learning-based techniques. Some notable methods include:
- ResNet: A deep residual network that has shown promise in modeling complex systems.
- Physics-Informed Neural Networks (PINNs): These networks incorporate physical laws into the learning process, providing a more accurate representation of the underlying dynamics.
- Autoencoders: Used for dimensionality reduction and feature extraction in complex datasets.
- Deep Operator Networks: These networks model the mapping between function spaces, making them suitable for solving differential equations.
Despite the promising results from these methods, the issue of solving stiff chemically reacting problems within the CFD framework remains significant. This study aims to address this issue by introducing the MLP-Mixer method named PatchTSMixer.
Research Methodology
Chemical Kinetic System
The ROBER system is a nonlinear coupled ODE system designed to model the chemical kinetics of an autocatalytic reaction. Due to the large discrepancy between the constants ( k_1 ), ( k_2 ), and ( k_3 ), the system presents considerable stiffness, making numerical integration nontrivial.
MLP-Mixer Method
PatchTSMixer is a relatively novel MLP-Mixer architecture intended to provide a lightweight alternative for time-series forecasting. The architecture splits multivariate time-series into small patches, which are then transformed by an intermediary layer into a multi-dimensional tensor. This tensor is subsequently passed through multiple MLP mixer layers, each learning correlations between patches and channels. The final stage reshapes the embedding to produce the output.
The model can be trained in two ways during the pretraining stage: as a masked autoencoder, suitable for multiple downstream finetuning tasks, and for direct forecasting, the approach preferred in this work. PatchTSMixer predicts a number ( h ) of future timesteps given a context length of size ( H ), which can be optimized during hyperparameter tuning.
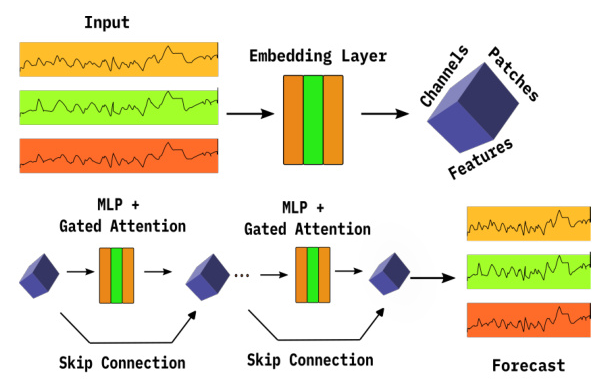
Experimental Design
Data and Training
The dataset used for the experiments was generated using the LSODA solver, designed to solve stiff problems. The ROBER system was integrated for ( t = 10^5 ) seconds from a randomly chosen initial state ([0.776, 6.913 \times 10^{-5}, 0.081]) and recorded with a resolution of ( \Delta t = 1 ) second. This initial condition was chosen to produce stiff chemical kinetics. The dataset was split into 60% for training, 20% for validation, and 20% for testing.
PatchTSMixer was trained using a context length ( H = 512 ), a prediction horizon ( h = 100 ), and a patch length ( p = 8 ). The model was trained for 300 epochs using the AdamW optimizer. After training, the model was used to forecast the testing region by small intervals to create the results.
Results and Analysis
To determine if PatchTSMixer can accurately capture and forecast stiff chemical kinetics, we compared its extrapolation predictions with the ground truth evolution obtained from the stiff numerical solver. This comparison is presented for the final 20,000 timesteps of the evolution.
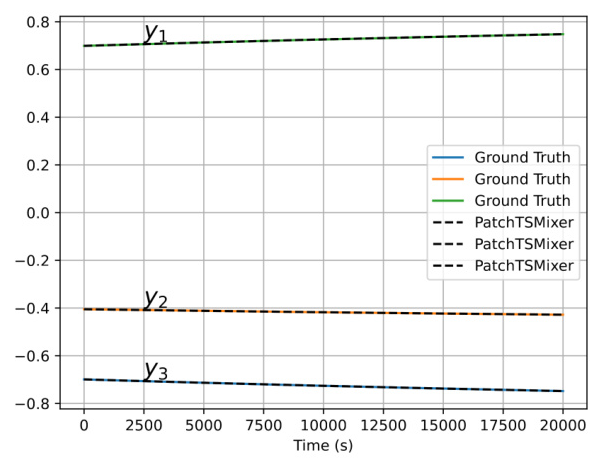
The extrapolation predictions of the chemical species ( y_i ) obtained from PatchTSMixer both quantitatively and qualitatively match the evolution from the numerical solver. The relative error for each extrapolation batch, corresponding to forecasts of 100 timesteps, showed a mean error of 0.0166% with a standard deviation of 0.0008%. However, PatchTSMixer was not as robust in dynamic extrapolation, where the output of a forecast batch is used to feed the next one, leading to deviations. This issue could potentially be alleviated through better choices of context and forecast dimensions.
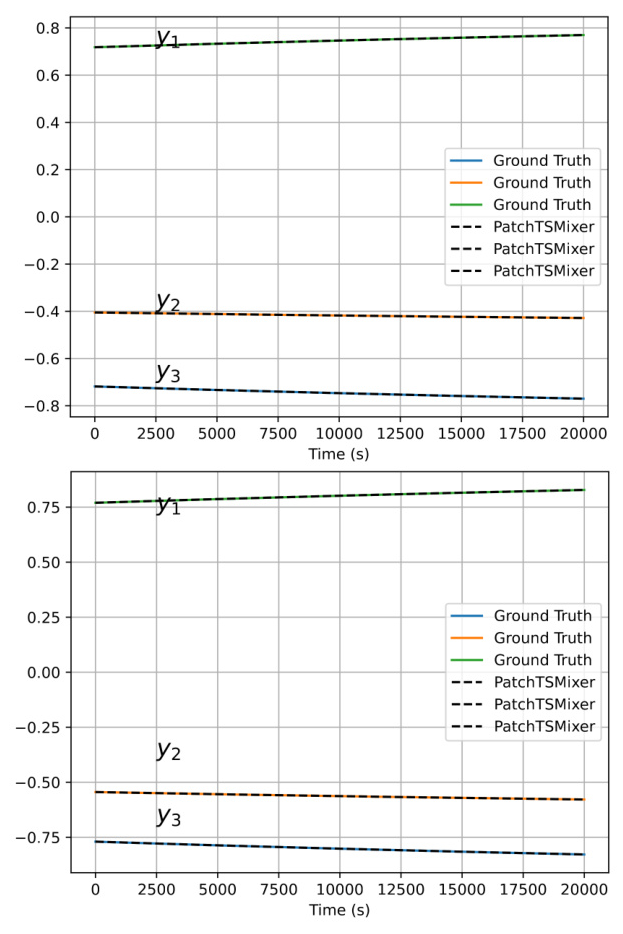
Overall Conclusion
In this study, we applied the recently proposed MLP-Mixer architecture, PatchTSMixer, in a novel SciML context to model the time-series evolution of stiff chemical kinetics, which is of significant industrial importance. Using the ROBER system, a benchmark model in chemical kinetics, we demonstrated a strong agreement between the evolution of the chemical species forecasted by PatchTSMixer and the ground truth. This result highlights the industrial utility of using an MLP-Mixer for forecasting the time-series evolution of stiff chemical kinetics and motivates its consideration as a building block for modern-day time-series foundation models.