

Authors:
Po-Yu Liang、Xueting Huang、Tibo Duran、Andrew J. Wiemer、Jun Bai
Paper:
https://arxiv.org/abs/2408.08341
Introduction
Peptides play a crucial role in various biological processes and have significant applications in drug discovery and biotechnology. Traditional methods for peptide discovery, such as sequence-based and structure-based approaches, often require extensive datasets, which can limit their effectiveness. This study proposes a novel method that utilizes autoencoder-shaped models to explore the protein embedding space and generate novel peptide analogs using protein language models. This method requires only a single sequence of interest, avoiding the need for large datasets.
Related Research
Lab Experiment Based Method
Traditional methods for identifying and optimizing peptide-based inhibitors include rational design, phage display, and directed evolution. These methods have limitations such as requiring extensive sequence and structure information, significant time and effort, and resources for screening.
Deep Learning Based Method
Recent studies have explored generating peptides with desired properties using deep learning methods. These methods focus on amino acid sequences and three-dimensional structures, utilizing models such as conditional variational autoencoders, generative adversarial networks, and Markov chain Monte Carlo methods.
Method
Hypothesis
The study hypothesizes that peptides with similar embeddings are likely to share higher property similarities, even if their sequence expressions differ. This hypothesis is inspired by word embedding studies, where vector abstract feature representations learned from deep learning models capture semantic meaning.
Definition
The proposed method employs an autoencoder-shaped model to learn the feature embedding. The dataset is defined as (X = {x_0, …, x_i, …, x_n}), where (x_i) is the amino acid sequence of a protein. The method is defined as (\hat{y}\tau = g(f(x_i) + \delta\tau)), where (\hat{y}\tau) is the generated amino acid sequence of the protein analog at step (\tau), (f(\cdot)) is a model projecting a protein sequence into the latent space, (\delta\tau) represents the noise added to the protein embedding at step (\tau), and (g(\cdot)) projects the noised embedding back to the sequence.
Overview of the Proposed Method
The proposed method involves three main steps: embedding, noise introduction, and decoding.
- Embedding: Projects peptide sequences from a discrete space into a continuous latent space.
- Noise Introduction: Introduces noise into the embeddings to explore the latent space systematically.
- Decoding: Converts the noised embeddings back into peptide sequences.
To validate the method, two embedding models were utilized: ProtT5 and ESM-2.
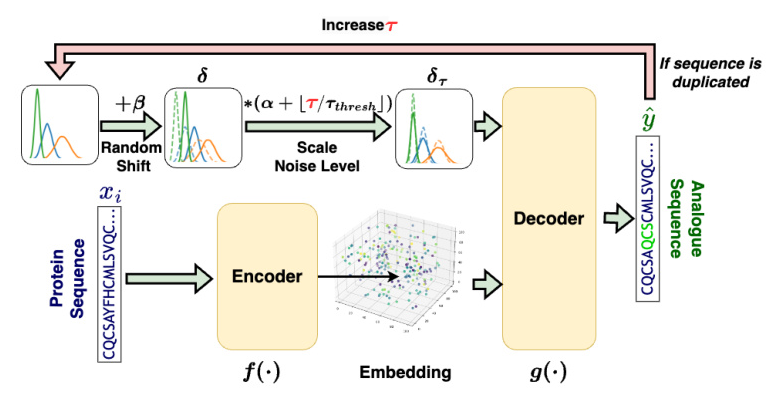
Embedding
Two state-of-the-art models, ProtT5 and ESM-2, were used to embed peptide sequences. Both models were pre-trained to leverage their advanced capabilities in understanding protein sequences.
- ProtT5 Embedding: Utilized the “Prot-T5-XL-Ur50” model, which provides an embedding size of 1024.
- ESM-2 Embedding: Utilized the ESM-2 model with 150 million parameters, providing an output size of 640.
Noise
Noise was introduced into the peptide embeddings to explore the latent space. The noise was drawn from a uniform distribution and adjusted to maintain its effectiveness. An adaptive approach was employed to balance finding sequences similar to and diverse from the original.
Decoder
Different functions were employed for ProtT5 and ESM-2 based on their distinct architectures.
- ProtT5 Decoder: Utilized a pre-trained decoder to project the transformed embeddings.
- ESM-2 Decoder: Trained a new decoder module, designed to be symmetric with the encoder’s architecture.
Data & Experiment Setup
Data Source and Filtering
The BioLip dataset was used to test the method. After filtering, the final dataset contained 4,758 unique peptide sequences. The UniProtKB/Swiss-Prot dataset was used to train the ESM-2 decoder module.
Baseline Models
Two baseline approaches were compared with the proposed method: random generated sequences and BLOSUM generated sequences.
Evaluation Metrics
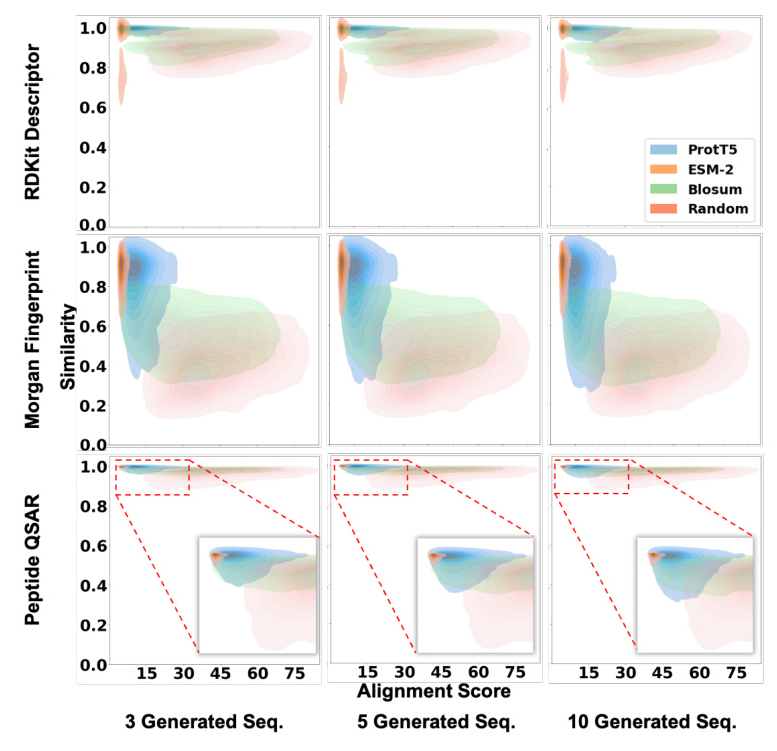
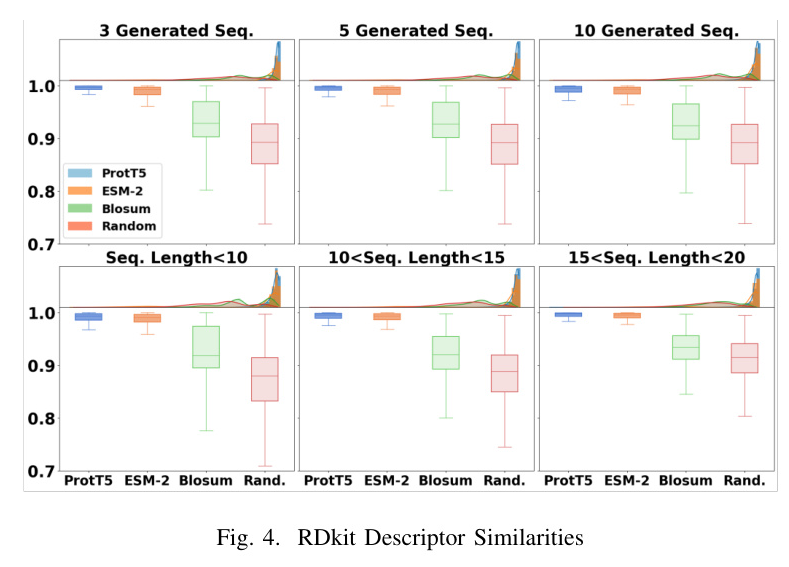
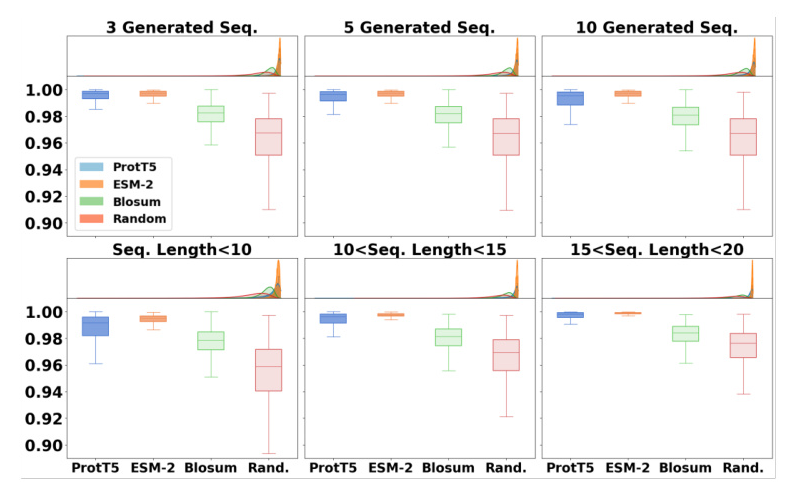
Three different indicators were used to evaluate the similarity between original and generated peptide sequences: Morgan Fingerprints, RDKit Descriptors, and QSAR descriptors.
Comparative Analysis
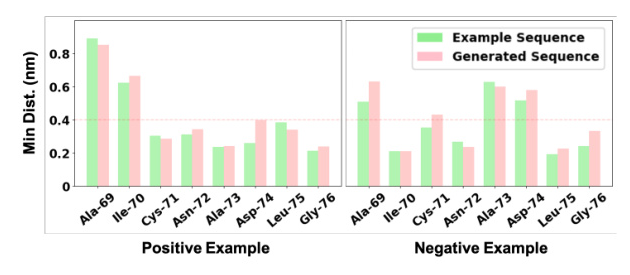
The method was applied to peptide ligands of the TIGIT receptor, identified through wet-lab experiments. Molecular Dynamics (MD) simulations were employed for further validation.
Result and Discussion
Overall Result
The proposed method outperformed baseline models in terms of average similarities for generating new sequences and peptides of different lengths. ProtT5 exhibited higher average similarity for RDKit descriptor similarity, while ESM-2 showed higher average similarity for Morgan fingerprint and sequence QSAR similarities.
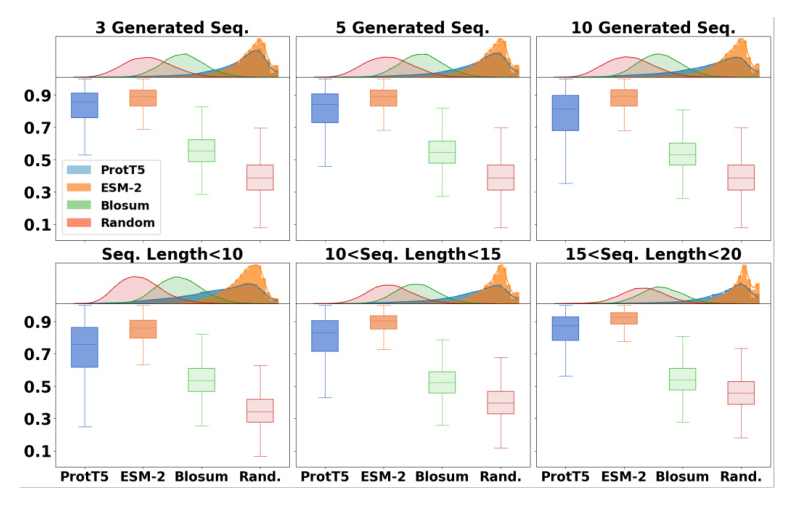
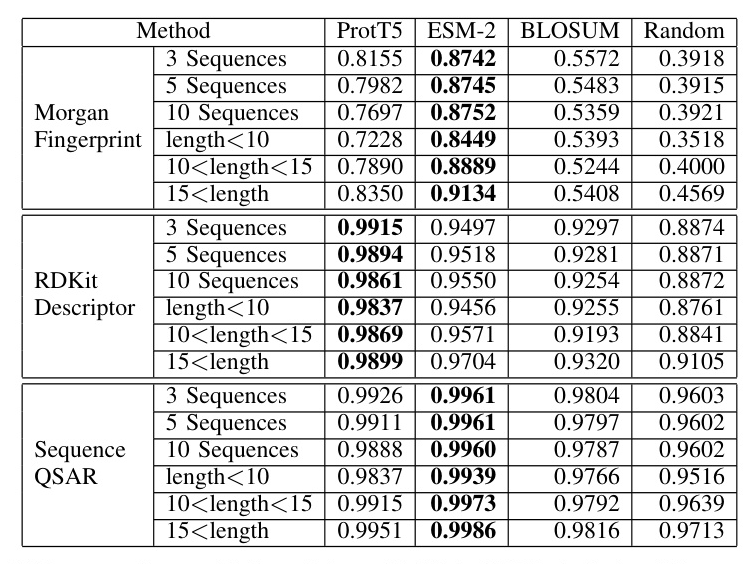
Physics Modeling Validation
MD simulations were used to validate the method with wet-lab experiments generated sequences. The generated sequences exhibited similar behavior to the original sequences, with comparable or improved binding affinity.
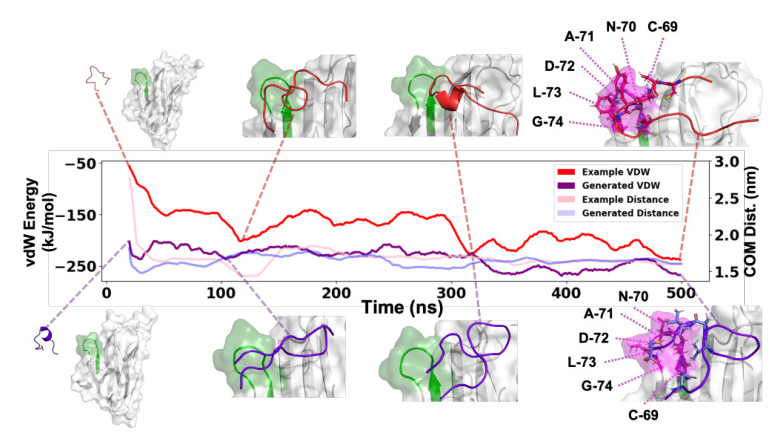
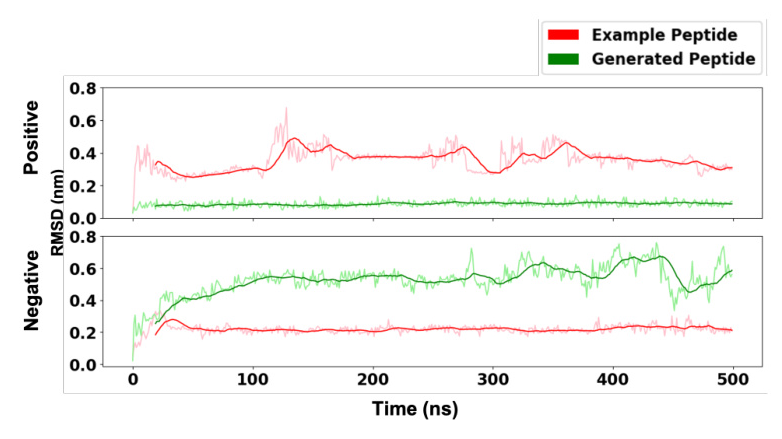
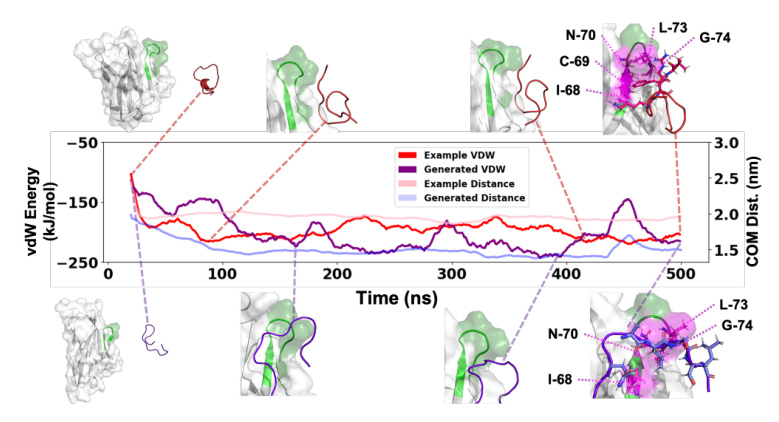
Conclusion
The proposed method addresses the challenge of generating peptides with desired properties by leveraging autoencoder models to explore the protein embedding space. The method significantly outperforms baseline models and demonstrates robustness through MD simulations. Future work will focus on testing the method in actual wet-lab experiments to further validate its effectiveness.
Code:
https://github.com/LabJunBMI/Latent-Space-Peptide-Analogues-Generation