

Authors:
Kamyar Zeinalipour、Neda Jamshidi、Monica Bianchini、Marco Maggini、Marco Gori
Paper:
https://arxiv.org/abs/2408.06396
Introduction
In recent years, the field of natural language processing (NLP) has achieved remarkable progress, particularly through the development and utilization of large pre-trained language models (LLMs). These sophisticated models represent a significant leap forward, primarily due to their ability to understand and generate human-like text based on training from extensive datasets. Typically, these models are trained using unsupervised learning techniques, where they learn to predict the next word or token in a sequence by examining the tokens that precede it. This method has propelled them to the forefront of various NLP applications, including chatbots, text summarization, and advanced information extraction tasks.
Among the intriguing avenues explored with these models is their application in the field of bioinformatics, specifically in protein generation. The primary objective of this research lies in advancing the understanding and application of medium-sized language models, particularly those in the 7 billion to 8 billion parameter range, including Mistral-7B, Llama-2-7B, Llama-3-8B, and gemma-7B, for the generation of high-quality protein sequences. Our hypothesis, backed by preliminary studies, suggests that these models, even when trained with considerably small datasets, can produce accurate and viable protein sequences effectively.
Related Works
The integration of NLP techniques into bioinformatics has transformative potential, particularly in the analysis of biological sequences such as DNA, RNA, and proteins. These biological data, sharing similarities with linguistic texts in their structured and functional building blocks, are highly amenable to computational methodologies. The impactful success seen in NLP through transformer-based models has led to breakthroughs in specialized models geared toward understanding the complexities of these biological sequences.
The realm of protein sequences has seen notable advancements through the adoption of both supervised and unsupervised learning models. Language models have been increasingly leveraged and employed in the domain of protein design. Supervised learning approaches refine models by training them with labeled data, which is invaluable for accurately predicting protein stability or identifying structural similarities among sequences. On the other hand, the introduction of transformer technology has been pivotal in popularizing unsupervised learning methods. These methods involve the strategic corruption of input sequences which are then used to train models to predict and reconstruct the natural sequence.
Methodology
From LLMs to Proteins
Large language models, such as transformers, are sophisticated algorithms trained on extensive textual datasets. These models utilize their predictive capability primarily to determine the subsequent token based on the preceding ones. Given their training on a vast amount of text data, LLMs are highly adaptable and can be finely tuned for specialized tasks, including summarizing specific document types like legal texts. An interesting application of these models is in the domain of protein generation. Proteins, being amino acid sequences, differ significantly from standard text data. This difference necessitates the retraining of tokenizers to achieve more accurate tokenization for proteins, enhancing the model’s ability to recognize and predict relevant patterns in amino acid sequences.
Tokenizer Retraining
In situations where the corpus significantly diverges from that utilized during the initial training of a language model, it becomes imperative to retrain the model from scratch. This process necessitates adjusting the tokenizer to accommodate the nuances of the new dataset. A tokenizer serves the critical function of converting textual data into numerical representations suitable for computational processing by language models. For the retraining of our tokenizer, we employed the Byte-Pair Encoding (BPE) method. BPE is a hybrid between a character-level and word-level tokenizer. It starts with a base vocabulary of individual characters and iteratively merges the most frequently adjacent pairs of characters or character sequences.
Fine-Tuning Pre-trained LLMs
In this research, our objective was to assess the capabilities of various pre-trained language models in the specialized task of protein generation. To this end, we fine-tuned four distinct models: Mistral-7B, Llama-2-7B, Llama-3-8B, and gemma-7B. Each model is based on the transformer architecture, which is renowned for its effectiveness in handling sequence-to-sequence tasks and operates under a causal framework conducive to generative tasks. The four models were specifically chosen to represent a bandwidth of computational capacities predominantly ranging between 7 billion and 8 billion parameters, enabling a focused analysis on how parameter scale influences model performance in biological sequence generation.
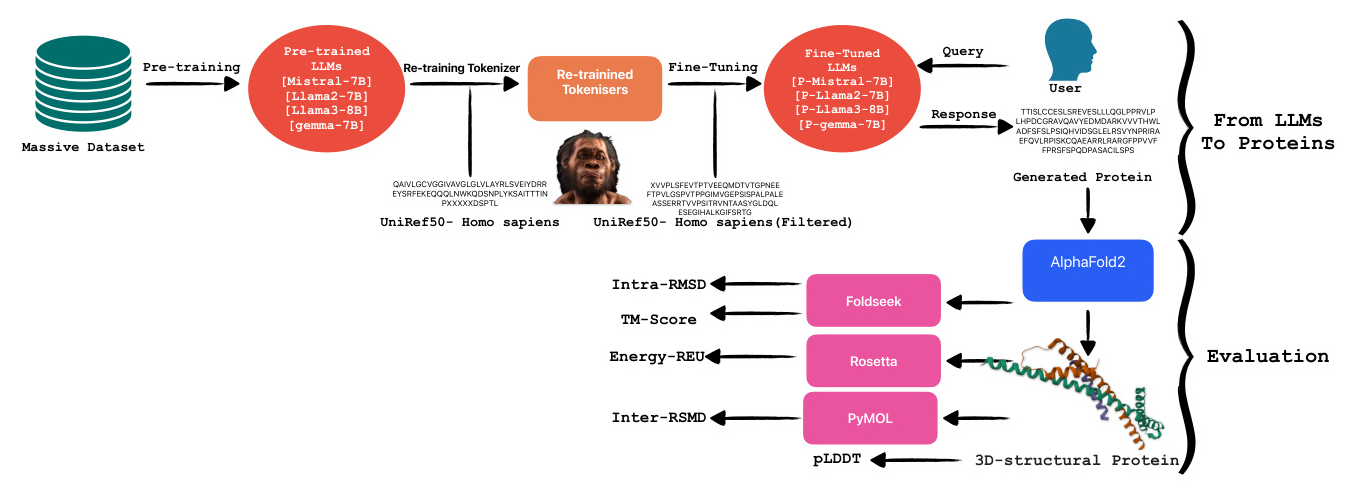
Evaluation
In this section, we describe each evaluation method implemented in our study following the generation of proteins. Initially, protein sequences generated using tuned LLMs were structurally modeled using AlphaFold2, which provided three-dimensional structures along with per-residue confidence scores (pLDDT). Subsequently, the topological similarity of these structures to known protein configurations was assessed using the TM-Score computed by FoldSeek. Additionally, Rosetta-Relax was employed to analyze the energetic profiles of the modeled proteins, enhancing our understanding of their stability and viability. For intra-dataset structural comparisons, RMSD calculations were conducted using PyMOL.
Alphafold2 (pLDDT)
In the initial phase of the evaluation, we utilized AlphaFold2 to predict the structures of the generated proteins and compute their predicted Local Distance Difference Test (pLDDT) scores. AlphaFold2, developed by DeepMind, represents a significant advancement in protein structure prediction by leveraging sophisticated deep learning methodologies. It predicts protein structures from amino acid sequences, using extensive training datasets of known protein structures and incorporating a self-attention mechanism.
Foldseek (TM-Score, Intra RMSD)
To evaluate the accuracy of predicted protein structures, we utilized Foldseek, a robust tool designed for the comparison and analysis of three-dimensional protein structures. By submitting our predicted protein models to Foldseek, we computed two critical metrics: the TM-score and Root Mean Square Deviation (RMSD). The TM-score, ranging from 0 to 1, quantifies the global topological similarity between two protein structures, with higher scores indicating greater structural resemblance. Conversely, RMSD is a widely used metric in structural biology that assesses the similarity between two protein structures by comparing the positional differences of corresponding atoms, typically those in the backbone, after optimal superimposition.
Rosetta-Relax (REU)
To comprehensively assess the quality of our predicted protein structures, we initiated the process by relaxing the native template. This initial relaxation ensures that the structure is energetically optimized from the outset, facilitating more accurate subsequent evaluations. Following the relaxation of the native template, we applied Rosetta-RelaxBB across all datasets. Rosetta-RelaxBB employs a Monte Carlo optimization approach that explores a range of backbone and rotamer conformations to minimize the Rosetta Energy function, which is based on biophysical principles and constraints.
PyMOL (Inter RMSD)
For the fourth phase of our evaluation, we utilized PyMOL, a sophisticated molecular visualization software equipped with extensive tools for protein structure analysis and comparison. PyMOL’s features facilitate detailed examination of molecular structures and enable various quantitative assessments, such as calculating the Root Mean Square Deviation (RMSD). Specifically, we determined the Inter RMSD, which quantifies the RMSD for each trajectory within our datasets.
Experimental Results
In this section, we delineate the experiments conducted in this study, presenting an evaluation of the results garnered from the protein sequences we generated. Additionally, we discuss the regeneration of proteins utilizing language-based models specifically designed for protein generation tasks, including ProGen in four distinct sizes, ProtGPT2, and ProLLaMA.
Dataset
In this study, the UniRef50 dataset, originating from the UniProt databases, has been utilized. The UniProt Reference Cluster (UniRef) databases systematically organize clustered sets of protein sequences from UniProtKB and selected UniParc records, aiming to reduce redundancy and provide comprehensive coverage of sequence space. Specific attention was given to the Homo sapiens subset within UniRef50, which initially comprised over 60,000 protein sequences. Given the constraints of computational resources and the criteria of our intended language models, a sequence length filter was applied. Only sequences below 512 tokens, as determined by our pre-trained tokenizer, were retained, narrowing the pool to 60,000 sequences.
Training Setup
The training methodology employed in this study involved training Language Models (LMs) specifically tailored for protein generation utilizing four Nvidia A6000 GPUs. The training configuration utilized a sequence length of 512, with a maximum training step limit of 2000 and a batch size of 1, coupled with a gradient accumulation step size of 16 for enhanced training efficiency. The learning rate was set at 5e-5, and a cosine learning rate scheduler was employed to adaptively adjust the learning rate. Furthermore, a weight decay of 0.01 and a num warm-up step value of 150 were applied to stabilize the training process. The utilization of the bfloat16 data format contributed to faster computation due to reduced precision, enhancing overall training performance.
Results Evaluation
In this section, we randomly selected 250 proteins, each with a length between 70 to 140 amino acids, from each of the under-investigation models for structure prediction and subsequent evaluation. In order to initiate the protein generation process, we input a special token, known as the beginning-of-sequence (BOS) token. Once this token is fed into the model, it begins to generate protein sequences, leveraging the patterns and knowledge it has acquired during its training phase. These proteins were submitted to AlphaFold2, which generated 3D structural models with corresponding pLDDT scores for each protein.
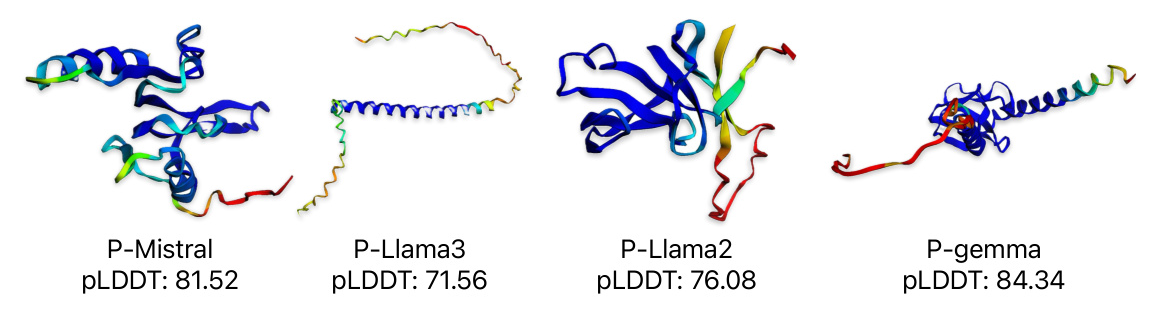
We proceeded to randomly select 20 3D structural proteins from each of the under-investigated models for a more in-depth analysis. The chosen proteins were then subjected to further evaluations, including the calculation of Intra RMSD, Inter RMSD, TM-Score, and REU with selected proteins. This multi-faceted approach to evaluation has allowed us to thoroughly assess the performance of our models and the quality of our 3D protein structure predictions.
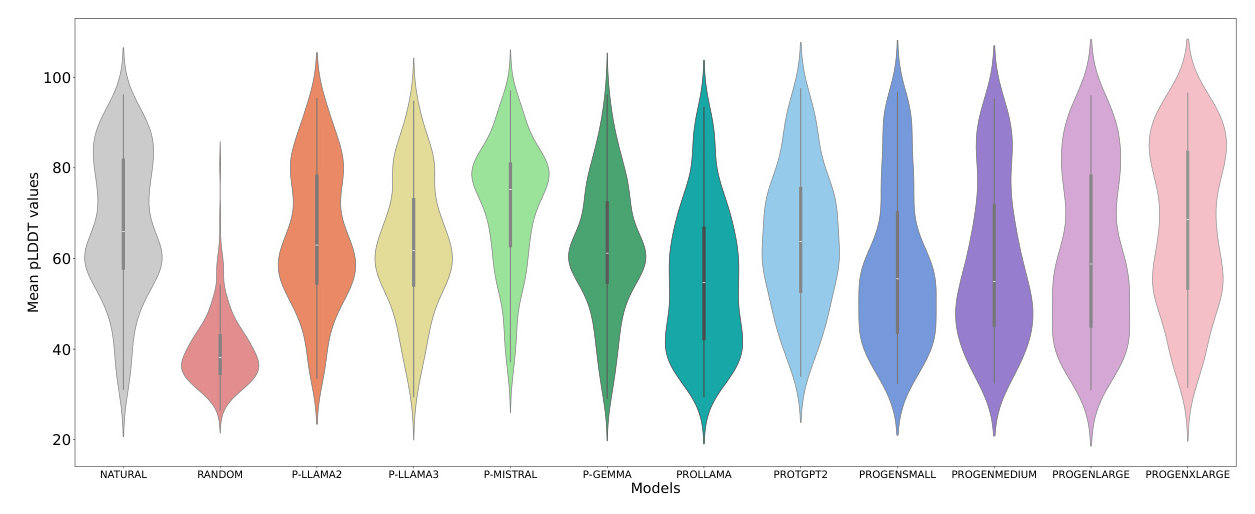
To evaluate the pLDDT score for each protein, AlphaFold2 generates five 3D structural models with corresponding pLDDT scores. We then calculated the mean of the five pLDDT scores to obtain a representative pLDDT score for each protein. We present the evaluation results using all the metrics discussed in Section 4. Table 1 summarizes the mean values of each evaluation metric for each model. Notably, P-Mistral consistently outperforms all other models across various metrics.
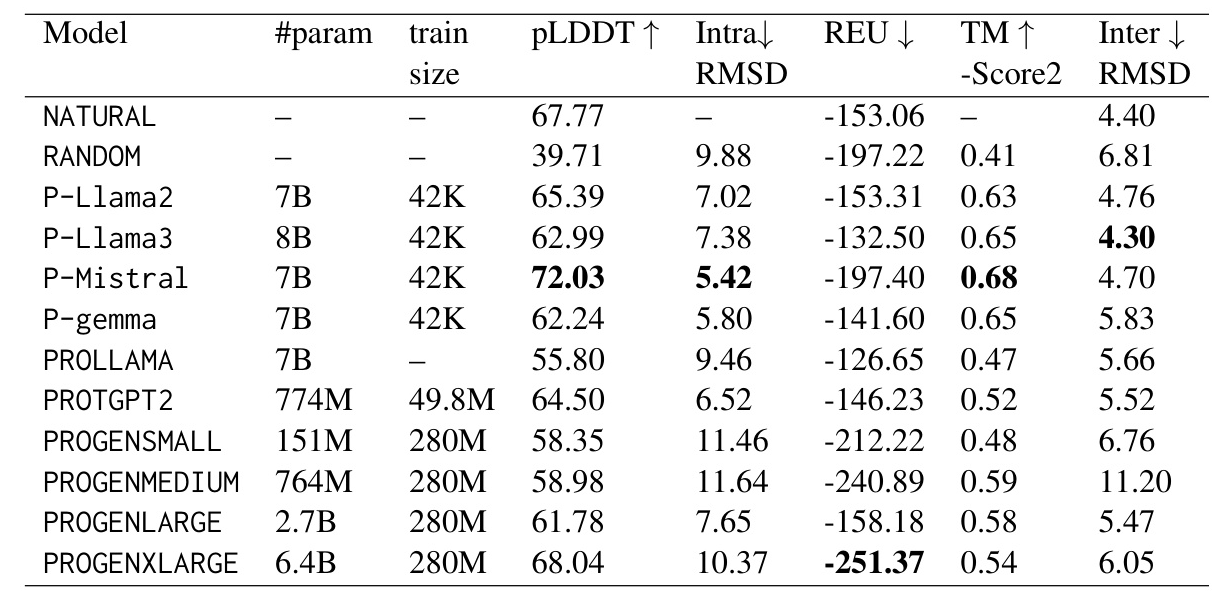
The most significant difference between the trained models and randomly generated proteins is observed in the pLDDT metric. Our models, P-Llama2 and P-Llama3, exhibit a distribution similar to the natural data. Additionally, we observed a significant disparity between randomly generated proteins and other models when evaluating the TM-score metric. Other metrics, such as Inter and Intra RMSD, are shown in Figures 8 and 6. Furthermore, for the REU metric, we identified an optimal range between -100 and -300. The randomly generated proteins fall significantly outside this interval, whereas the models we introduced predominantly fall within the same range as the natural data.
Conclusion
In this study, we introduced four novel models designed to generate high-quality protein sequences by leveraging pre-trained language models. This research is motivated by the growing demand for efficient and accurate tools that can assist in understanding and engineering protein structures, which are pivotal in numerous biological and medical applications. Our approach involved a meticulous design and training phase, followed by rigorous testing and validation processes to assess the performance of each model.
To provide a thorough evaluation, we conducted comprehensive experiments comparing our models with a range of existing models that also utilize language models for protein sequence generation. Comparative analyses were performed, which were grounded on diverse criteria, including sequence quality, diversity, and fidelity to biological functions. These analyses also incorporated several structural assessment metrics such as pLDDT (predicted Local Distance Difference), TM-Score (to assess structural similarity), RMSD (Root Mean Square Deviation), and REU (Rosetta Energy Unit).
Our findings revealed that some of our proposed models, particularly P-Mistral, exhibited superior performance compared to existing models, even surpassing those trained on considerably larger datasets. This remarkable performance underscores the potential of our models to offer significant advancements in the field of protein sequence generation. We are committed to the principles of open science and reproducibility. Consequently, we will make all four models publicly available to the research community. This accessibility will empower other researchers to utilize and build upon our work, fostering further advancements in the field of protein sequence generation.
Moreover, we aim to extend the capabilities of these models by incorporating instruction tuning to generate proteins with specific constraints. This will involve refining the models to adhere to certain criteria, such as ensuring the sequences have particular structural or functional properties. Such advancements could be pivotal in various applications, including drug design, synthetic biology, and understanding protein interactions at a deeper level. While our current implementation of LLMs for protein generation excels in unconditional generation, there is a need to explore and develop methods for generating conditional proteins. This would allow us to guide the generation process toward specific protein characteristics or functions, thereby enhancing the practical utility of our model.
Acknowledgments
The funding for this paper was provided by the TAILOR project and the HumanE-AI-Net projects, both supported by the EU Horizon 2020 research and innovation program under GA No 952215 and No 952026, respectively.